
Amyotrophic Lateral Sclerosis (ALS) is a relentless and complex neurodegenerative disorder that affects the central nervous system, gradually impairing voluntary muscle movement. Referred to as Lou Gehrig’s disease, ALS has fascinated the scientific and medical communities due to its perplexing nature.
ALS is characterized by the degeneration of motor neurons responsible for transmitting signals from the brain to the muscles throughout the body. This progressive loss of motor neurons leads to muscle weakness, paralysis, and eventually respiratory failure. Patients experience a range of symptoms, including muscle cramps, muscle twitching (fasciculations), difficulty swallowing (dysphagia), slurred speech (dysarthria), and muscle stiffness (spasticity). The relentless progression of ALS presents immense challenges for those affected, necessitating a deeper understanding of the disease.
ALS is a global health concern, affecting people of all ethnicities and geographic regions. According to a systematic review and meta-analysis [1], the overall crude worldwide incidence of ALS was 1.59 per 100,000 person-years. The prevalence of ALS increases with age, with the majority of cases diagnosed between the ages of 40 and 70.
Currently, ALS treatment therapies primarily focus on managing symptoms and improving patients’ quality of life. There are three approved medications: riluzole, edaravone, and Relyvrio, two of which have shown moderate efficacy, resulting in a slight increase in survival of several months [2, 3]. Additionally, supportive therapies such as physical and occupational therapy, speech therapy, and assistive technologies help maintain functionality and autonomy. It is important to note, however, that these therapeutic options do not halt or reverse the progression of the disease. This underscores the critical importance of exploring novel techniques to address ALS’s relentless progression in the fight against the disease.
While the majority of ALS cases develop spontaneously, approximately 5-10% of cases exhibit familial patterns, suggesting an underlying genetic predisposition. The identification of SOD1 (superoxide dismutase 1) as the first causal gene for ALS dates back to 1993 [4]. Since then, a number of genes, including C9orf72, TARDBP, FUS, and others, have been linked to the pathophysiology of ALS. These genes play crucial roles in protein regulation, RNA metabolism, and cytoskeleton organization, among other cellular functions. By elucidating the genetic basis of ALS, we not only gain a better understanding of the disease but also open up new avenues for targeted therapeutics and personalized medicine, paving the way for more precise interventions.
In order to gain a better understanding of ALS pathogenesis and develop effective treatments, mouse models play a crucial role. GemPharmatech has developed a proprietary mouse model named B6-hSOD1 G93A, which carries the full-length human SOD1 gene with the G93A mutation, including the human promoter sequence, exons, and introns. This model proves invaluable in evaluating ASO drugs targeting the SOD1 pathway and other ALS therapies. Preliminary validation results demonstrate that this mouse model partially replicates the clinical and behavioral characteristics observed in ALS patients, with disease onset occurring at approximately 5.5 months of age. As a result, it serves as an ideal research tool for investigating ALS pharmacology, drug efficacy, and disease mechanisms.


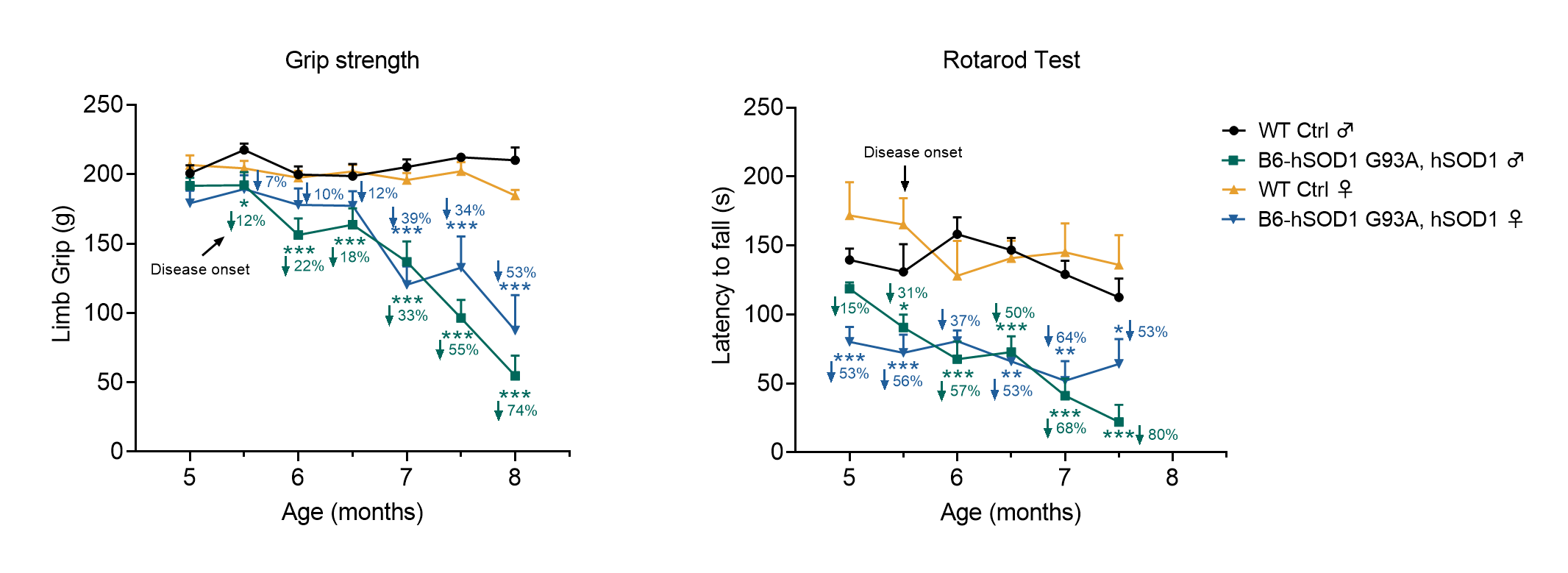
Figure 1. Muscle histopathology of B6-hSOD1 G93A, hSOD1 male mice
Figure 2. Motor capability changes in B6-hSOD1 G93A, hSOD1 mice

Figure 3. Transcriptome expression profile changes in the spinal cord of B6-hSOD1 G93A, hSOD1 mice at the disease onset
Aside from the well-studied SOD1 gene, GemPharmatech has successfully developed a proprietary mouse model, known as B6-hTDP43 A315T, which specifically targets another crucial gene associated with ALS, TARDBP. This model proves to be an invaluable resource for comprehending the involvement of the TARDBP gene or TDP43 protein in ALS, as well as for evaluating drug effectiveness and elucidating disease mechanisms.
To discover further information about our research and obtain a complimentary quote, we invite you to visit our website today.
References:
- Xu L, Liu T, Liu L, Yao X, Chen L, Fan D, Zhan S, Wang S. Global variation in prevalence and incidence of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol. 2020 Apr;267(4):944-953.
- Jaiswal, M.K., Riluzole and edaravone: A tale of two amyotrophic lateral sclerosis drugs. Med Res Rev, 2019. 39(2): p. 733-748.
- Chio, A., L. Mazzini, and G. Mora, Disease-modifying therapies in amyotrophic lateral sclerosis. Neuropharmacology, 2020. 167: p. 107986.
- Deng, H.X., Hentati, A., Tainer, J.A., et al. (1993). Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 261, 1047–1051.
It’s wonderful to see thoughtful discussions about supportive care for loved ones. Small acts of companionship can brighten days, reduce isolation, and promote a sense of normalcy for seniors and caregivers alike companion care services.
Thank you for sharing this thoughtful post. It’s reassuring to learn about practical options that support families, caregivers, and loved ones alike while maintaining dignity and comfort at home respite care services.
I appreciate how informative and supportive this post is for anyone dealing with concerns about thinning or shedding hair. Clear tips and compassionate guidance can make a real difference in taking the next steps hair loss specialist appointment in Bertrange.
Great post—really helpful insights into staying active and motivated. I appreciate practical tips that fit into a busy schedule and encourage consistency without overdoing it. Thanks for the balanced guidance mens cardio.
As a longtime reader, I appreciate thoughtful insights that blend practical tips with a friendly, practical vibe. The perspectives here are refreshing and genuinely helpful for any reader seeking clear guidance Educational psychologist Cape Town.
As a reader, I appreciate practical tips and clear guidance on foot comfort. Small changes can make a big difference for daily walking and overall posture. Thanks for sharing thoughtful insights feet orthotics near me.
This post really highlighted practical tips for maintaining a bright smile and healthy gums, and I appreciate the friendly, informative approach that makes dental care feel approachable for everyone involved Dentist Conroe Texas.
This thoughtful post highlights the value of open communication and patience in relationships, offering practical tips that resonate with couples seeking understanding, growth, and a stronger connection through mindful, collaborative steps Couples Counselling Markham.
I found this post thoughtful and reassuring, offering practical ideas and a compassionate tone. The tips feel useful for readers seeking gentle, evidence‑based approaches to healing and personal growth after difficult experiences Hypnotherapy for Trauma Support.
That insights shared really resonated with me, especially the emphasis on compassionate support and flexible plans that suit families navigating care needs with dignity and respect Coquitlam Home Care Services.
Appreciate this thoughtful post on recovery journeys after injuries; sharing practical tips and compassionate guidance helps communities stay informed and motivated to seek timely care and personalized rehabilitation Motor Vehicle Accident Physiotherapy Langley.
I appreciate the clear, practical advice shared here about foot care and treatment options. It’s reassuring to see compassionate guidance and evidence-based tips that help readers feel more confident about seeking care when needed nail surgery near me.
Love how this post emphasises communication and empathy as foundations for stronger bonds; practical tips and real-world examples make it easy to apply advice beyond the page. Thanks for sharing thoughtful insights relationship therapy abbotsford.
Appreciate this thoughtful discussion. It’s reassuring to see clear, practical information and personal stories that help people feel informed and supported when navigating sensitive medical choices Vasectomy Removal in Southern California.
This thoughtful post highlights the importance of a balanced approach, combining evidence-based medical care with supportive, compassionate wellbeing practices that empower patients on their healing journey and improve quality of life Holistic Cancer Treatment Florida.
Great post and thoughtful points here. I appreciate practical tips and the supportive tone that makes fitness feel approachable for everyone, whether you’re new or returning after a break Bootcamp Hallett Cove.
Great insights in this post; it really highlights how patient understanding and compassionate guidance can make a big difference when navigating treatment options and potential risks together with healthcare providers medical marijuanna doctors.
I found the insights calming and thoughtful, offering practical tips that feel achievable in daily life. The reflections on pacing, breath, and mindful moments resonate deeply and invite gentle self‑care Silent Meditation Retreat Arizona.
Great points raised here about convenience and access. With flexible options and patient-centered care, remote consultations can support timely advice and ongoing support for families navigating busy schedules Telehealth Services in QLD.
I appreciate this thoughtful post and agree that choosing a reputable clinic and discussing goals with a qualified professional is key to safe, satisfying results. A calm, informed approach makes all the difference Affordable botox clinics near me.
Really appreciate this thoughtful post—love how it highlights practical steps for mindfulness and calm. It’s refreshing to see simple techniques that fit into a busy lifestyle without feeling overwhelming deep breathing exercises app.
I really appreciate this insightful post—clear, practical tips that feel achievable. It’s reassuring to see realistic expectations and gentle routines that fit into busy days, without jumping to drastic choices Under Eye Bags Treatments.
This post highlights how thoughtful, patient-focused care can make a real difference. A calm, collaborative approach helps families feel supported and confident in their health decisions over time Concierge Medicine Rockville.
This thoughtful piece really resonated with readers seeking practical tips and reassurance. I appreciate the balanced guidance and relatable examples that make healthy choices feel achievable for busy lifestyles kotex cooling pad.
Great post—very insightful and balanced. I found the tips practical and accessible, especially for someone weighing cosmetic options. It’s reassuring to see clear guidance on choosing qualified clinicians and realistic expectations liposuction chin malaysia.
Grateful for insightful guidance on coping after medical care. This discussion highlights practical steps, compassionate support, and practical tips that make recovery feel supported and achievable for anyone facing a transition home Post Hospital Care services.
What a thoughtful post—everyday health basics like quick responses, staying calm, and calling for help can truly save lives during emergencies. Thanks for sharing practical, accessible advice for readers near and far Heart Attack Care Lagos.
I appreciate the thoughtful discussion here and found practical tips that make choosing a solution easier. It’s reassuring to hear about careful planning, durable results, and ongoing care from trusted professionals dental implants near me.
Appreciate the thoughtful post on maintaining oral health; it’s always encouraging to hear practical tips, gentle reminders about routine care, and the value of seeking professional guidance for any concerns restorative dental services.
Great post with practical tips—I’ve found small daily habits add up over time, keeping motivation steady. Consistency beats intensity, and a supportive routine makes workouts feel more enjoyable and sustainable NYC Personal Trainer.
I really appreciated the thoughtful discussion here and found the insights helpful for considering everyday wellness. Thanks to everyone for sharing experiences and practical tips that feel doable for busy readers SS-31.
Me parece muy interesante la labor que realiza la Clínica González Gayoso en el ámbito de la salud. Su compromiso con la atención personalizada y el bienestar integral de los pacientes es fundamental para mejorar la calidad de vida. Sin duda, la Clínica González Gayoso es un referente importante en